18 How a neuron is excited
Gradients, Channels, Synaptic Potentials, and the Action Potential
The previous chapter introduced the main cell types: neurons and glia, the shapes neurons take, and the tripartite synapse formed with a pre- and post-synaptic neuron and astrocyte. We ended by discussing circuits — a neuron influences its target by excitation or inhibition — and then promising to explain how that influence actually happens. Here, we discuss that fast electrical layer of signaling: the millisecond machinery by which one neuron excites or inhibits another.
The unit’s organizing claim is that signaling happens across many timescales, and that the fast electrical layer is the most recent, most specialized, and most expensive in terms of energy use. We previewed the reason in the overview: an animal cell, having given up the rigid wall for the freedom to move, must pump ions across its membrane for its whole life simply to maintain its state, and those pumped gradients are a store of energy that the neuron later learned to spend in quick, controlled bursts. The single thread running through all of this chapter is the interaction of two gradients — a chemical gradient and an electrical gradient — acting on the same ions at the same time. Almost everything that follows is a consequence of those two forces, sometimes cooperating and sometimes opposed, and of the channels that decide, moment to moment, which ions are allowed to cross the cell’s membrane.
We will build the story in order. First the two gradients and the equilibrium that results when they balance, for a single ion (the Nernst equation) and then for the real membrane with several ions at once (the Goldman–Hodgkin–Katz equation). Then the disequilibrium that this creates — a membrane poised like a loaded spring — and the pump that keeps it loaded. Then the gated channels that release it: the locked gate, the doorbell, and the emergency door. Then the small, local, graded events these produce at synapses (post-synaptic potentials), how a neuron adds them up (summation), and finally the large, regenerating, all-or-none event that carries the result down a long axon (the action potential), together with the myelin that makes it fast. Along the way we will meet the people who worked this out — Walther Nernst, David Goldman, Alan Hodgkin and Andrew Huxley, Bernard Katz — because the concepts are easier to remember when attached to the problems their discoverers were trying to solve.
18.1 Two gradients acting on one ion
Begin with the simplest possible question. An ion sits in solution on one side of a membrane. Which way will it move, and why? There are exactly two reasons an ion moves, and keeping them separate is the key to the whole chapter.
The first is the chemical gradient, also called the concentration gradient. A dissolved substance tends to spread from where it is crowded to where it is sparse. This is just diffusion, the same process by which a drop of ink disperses through a glass of water or a smell fills a room. There is nothing mysterious in it: molecules are in constant random motion, and random motion carries more of them out of a crowded region than into it, until the crowding is even everywhere. At that point motion has not stopped — individual molecules still jostle in every direction — but there is no longer any net movement in one direction. That balanced condition is equilibrium. An ion, being a dissolved substance, feels this concentration force like any other: if there is more sodium outside the cell than inside, sodium tends to diffuse in.
The second reason is the electrical gradient. An ion, unlike a drop of ink, carries a charge, and charge responds to charge: opposite charges attract, like charges repel. If the inside of a cell is electrically negative relative to the outside, a positive ion will be pulled inward toward the negativity, and a negative ion will be pushed outward away from it, regardless of how the concentrations stand. (A point of terminology that has tripped up generations of students, including the lecturer whose course these chapters follow: a positively charged ion is a cation and a negatively charged one is an anion, and the names run counter to intuition because cations move toward the negative electrode, the cathode, and anions toward the positive one, the anode. The chemistry is sensible; only the vocabulary is confusing.)
Here is the crucial move, and the thread of the chapter: these two forces act on the same ion at the same time, and they need not agree. The chemical gradient might push an ion one way while the electrical gradient pulls it the other. To know what the ion will actually do, you cannot consult either gradient alone; you have to combine them. Their combination is the electrochemical gradient, and the net push it produces on an ion is the electrochemical driving force. When chemists and physiologists speak of an ion “wanting” to move, this combined force is what they mean — a convenient shorthand, never a claim that ions have desires.
A few ions do almost all the work in neurons: sodium (\text{Na}^+), potassium (\text{K}^+), chloride (\text{Cl}^-), and calcium (\text{Ca}^{2+}), with magnesium (\text{Mg}^{2+}) making a cameo later when we discuss the NMDA receptor in Chapter 20. Sodium and potassium are the protagonists of this chapter. Their distributions across the membrane are nearly mirror images of each other, and that asymmetry is the raw material of every fast signal a neuron sends.
The asymmetry is worth seeing as numbers, because the rest of the chapter is, in a sense, a long commentary on this one table. The values below are representative concentrations for a typical mammalian neuron; exact figures vary by cell type, region, and method, so treat them as orders of magnitude rather than constants of nature. The unit mM means millimolar, or millimoles of a substance per liter of solution; 1 mM is one-thousandth of a mole per liter.
| Ion | Inside (mM) | Outside (mM) | Gradient points | Equilibrium potential |
|---|---|---|---|---|
| Sodium (\text{Na}^+) | ~10–15 | ~145 | inward | \approx +60 mV |
| Potassium (\text{K}^+) | ~140 | ~4–5 | outward | \approx -90 mV |
| Chloride (\text{Cl}^-) | ~5–10 | ~110–120 | inward | \approx -70 mV |
| Calcium (\text{Ca}^{2+}) | ~0.0001 | ~1–2 | inward | \approx +120 mV |
| Magnesium (\text{Mg}^{2+}) | ~0.5–1 | ~1–1.2 | inward (weakly) | shallow |
Read the table with the two gradients in mind and the whole chapter is foreshadowed. Sodium is crowded outside and will be drawn toward the negative inside, so both of its gradients point the same way and its equilibrium potential is strongly positive — sodium is the loaded spring. Potassium is the mirror image, crowded inside, with a deeply negative equilibrium potential. Chloride sits with its equilibrium potential close to the resting potential we are about to derive, which is why it will turn out to inhibit so gently. And magnesium is the odd one: its gradient is shallow, almost flat, so it is not a signal-carrier in the way the others are — which is exactly what will make it useful later as a voltage-sensitive plug rather than a current.
One caution about the last two rows. For sodium, potassium, and chloride, the concentration in the table is essentially the free, dissolved ion, because these ions are mostly free in solution. Calcium and magnesium are different. The free intracellular calcium is the ~0.0001 mM listed, but much larger quantities are bound to proteins or sequestered in intracellular stores, especially the endoplasmic reticulum in neurons and glia. The free intracellular magnesium is the ~0.5–1 mM listed, but the total intracellular magnesium is far higher — on the order of 14–20 mM — because most of it is bound to ATP and other phosphate compounds or sequestered in organelles. It is the free ion that matters for the membrane, so that is what the table reports, but the discrepancy is a reminder that “concentration” in a living cell is not always a simple thing to state. We will not need magnesium again until we reach the NMDA receptor in the chapter on plasticity; it is in the table only so that the cast is complete.
18.2 Equilibrium for a single ion: the Nernst equation
Let us make the two-gradient idea concrete with the cleanest possible case. Imagine a membrane with potassium crowded on the inside and essentially none on the outside, and imagine that the only thing trapped inside along with the potassium is a population of large negatively charged molecules — proteins and nucleic acids — far too big to pass through any pore. (This is not an artificial setup; the inside of a real cell is genuinely full of such large anions, for the simple reason that the molecules of life are mostly big and mostly negative.) Now punch holes (Figure 18.2, leak channels) in the membrane just wide enough for potassium to pass.
What happens? At first, potassium does what any crowded substance does: it diffuses outward, down its concentration gradient, toward the empty outside. But each potassium ion that leaves carries a positive charge with it, and it leaves behind one of those big negative anions that cannot follow. So with every ion that exits, the inside grows a little more negative relative to the outside. That growing negativity is an electrical gradient, and it pulls the positive potassium back in. The two forces now oppose each other: the concentration gradient pushing potassium out, the electrical gradient — which the outflow itself created — pulling it back.
The outflow does not continue until the concentrations are equal. It stops well short of that, at the point where the inward electrical pull exactly cancels the outward chemical push. There, once again, individual ions cross in both directions, but there is no net movement. The membrane voltage at which this balance occurs, for a single ion species, is that ion’s equilibrium potential (also called its Nernst potential). It is the voltage that exactly offsets the ion’s concentration gradient — the electrical bribe required to make the ion content to stay put.
We can calculate it. The relationship was worked out by the German physical chemist Walther Nernst in the 1880s, and it is named for him. The Nernst equation looks more intimidating than it is:
E_{\text{ion}} = \frac{RT}{zF} \ln \frac{[\text{ion}]_{\text{out}}}{[\text{ion}]_{\text{in}}}
Strip away the constants and the heart of it is a single ratio: the concentration of the ion outside the membrane divided by its concentration inside. R is the gas constant and T is the absolute temperature — together they capture the fact that diffusion is driven by thermal motion, so the force depends on how warm the system is. F is Faraday’s constant, which simply converts between amounts of charge and amounts of substance. And z is the valence of the ion — its charge with sign, +1 for potassium or sodium, -1 for chloride, +2 for calcium — which is what makes the equation come out with the correct polarity for negative versus positive ions. The natural logarithm appears because the relationship between a concentration ratio and a voltage is logarithmic, not linear; but you do not need to understand the logarithm to use the result. You do not even need to compute it by hand. Any number of online calculators will solve it. What matters is the concept: the equation converts a concentration ratio into the voltage that balances it.
Run the numbers for potassium, using concentrations typical of a mammalian neuron — roughly 140 millimolar inside and 5 millimolar outside — and the equilibrium potential comes out near -90 to -97 mV. The sign tells the story we just narrated in words: to keep potassium from leaving a cell in which it is so heavily concentrated, the inside must be held strongly negative. That negativity is the electrical price of potassium’s concentration gradient.
Now do the same for sodium, whose distribution is nearly the reverse — about 145 millimolar outside and only 5 to 15 millimolar inside. Sodium’s equilibrium potential comes out around +60 mV, positive. Again the sign is doing real work. Sodium is crowded outside and would diffuse in; to hold it out, you would have to make the inside strongly positive to repel it. The contrast between potassium’s deeply negative equilibrium potential and sodium’s strongly positive one is not a numerical curiosity. It is the loaded spring of the whole system, and we will return to it in a moment.
If you would like to see why the equation has the shape it does, here is the intuition behind each piece.
The logarithm of a concentration ratio appears because the entropic tendency to spread out depends on relative crowding, not absolute numbers. Doubling both concentrations changes nothing about which way diffusion pushes; only the ratio matters, and the natural measure of a ratio’s “size” in thermodynamics is its logarithm.
The factor RT scales the whole thing by thermal energy. Diffusion is just thermal jostling, so the harder the molecules are jostling (the higher the temperature), the larger the voltage needed to hold them against their concentration gradient. At body temperature, RT/F works out to about 26–27 mV, and if you convert from natural log to base-10 log the prefactor becomes the famous \sim 61 mV that appears in many textbooks: E_{\text{ion}} = \frac{61\,\text{mV}}{z}\log_{10}\frac{[\text{ion}]_{\text{out}}}{[\text{ion}]_{\text{in}}} at 37°\text{C}.
The valence z in the denominator handles charge. A divalent ion like calcium (z = +2) carries twice the charge per ion, so a given concentration ratio is balanced by half the voltage. A negative ion like chloride (z = -1) flips the sign, which is exactly right: for an anion, the polarity that balances a given gradient is reversed.
None of this is required to follow the rest of the chapter. Just remember that the Nernst equation turns a concentration ratio into the voltage that balances it, for one ion in isolation.
18.3 The resting membrane: the Goldman–Hodgkin–Katz equation
The Nernst calculation answers a question no real neuron actually faces, because no real membrane has only one ion crossing it. A living neuron’s membrane is permeable to several ions at once — potassium and sodium and chloride all have routes across it — and each of them has its own, different equilibrium potential. They cannot all be satisfied simultaneously. If the membrane sits at potassium’s equilibrium potential of -90 mV, then sodium, whose equilibrium potential is +60 mV, is wildly out of balance and floods inward; if it sits at sodium’s +60 mV, potassium floods out. The membrane has to settle somewhere, and where it settles is a compromise. We call that compromise the resting membrane potential.
Two things determine the compromise. The first is each ion’s equilibrium potential, which we already know how to compute. The second is new and decisive: the membrane’s permeability to each ion — how easily that ion can actually get across. Permeability is not the same as the concentration gradient. An ion can have an enormous gradient and yet contribute almost nothing to the resting voltage if the membrane gives it almost no way through. And here is the central physical fact about a resting neuron: its membrane is studded with potassium channels that are simply always open — leak channels — and has very few open sodium channels by comparison. The membrane is roughly twenty to thirty times more permeable to potassium than to sodium at rest. So in the tug-of-war over the resting voltage, potassium pulls with far more leverage, and the resting potential ends up much closer to potassium’s equilibrium potential than to sodium’s.
The equation that combines all of this is the Goldman–Hodgkin–Katz equation, usually shortened to the GHK or Goldman equation, after David Goldman, who published it in 1943, and Alan Hodgkin and Bernard Katz, who applied it to nerve. It is the Nernst equation grown up: instead of one ion it includes a term for each, and instead of bare concentrations it weights each concentration by that ion’s permeability, written P.
V_{m} = \frac{RT}{F} \ln \frac{P_{\text{K}}[\text{K}^+]_{\text{out}} + P_{\text{Na}}[\text{Na}^+]_{\text{out}} + P_{\text{Cl}}[\text{Cl}^-]_{\text{in}}}{P_{\text{K}}[\text{K}^+]_{\text{in}} + P_{\text{Na}}[\text{Na}^+]_{\text{in}} + P_{\text{Cl}}[\text{Cl}^-]_{\text{out}}}
Do not be alarmed by its size; it is the same idea repeated. Three points are worth noticing. First, each ion enters weighted by its permeability P, so an ion the membrane barely admits contributes little no matter how steep its gradient — permeability is the volume knob on each ion’s vote. Second, chloride appears with its inside and outside concentrations swapped relative to the positive ions; this is the bookkeeping that accounts for its negative charge, and it lets the equation handle anions and cations in one expression. Third, there is no valence term out front as there was in the Nernst equation, because the swap-and-sum handles the signs internally; the GHK equation as written assumes the ions are singly charged, which for potassium, sodium, and chloride is fine.
Now watch what the equation tells us. Plug in the real permeabilities — set potassium’s to 1 as a reference, sodium’s to about 1/20 of that, chloride’s to somewhere in between — along with the real concentrations, and solve. The answer comes out near -65 to -70 mV. This is the resting membrane potential of a typical neuron, the voltage you would measure with one electrode inside the cell and one outside while the neuron sits quietly. As a check on the logic, set sodium’s and chloride’s permeabilities to zero, leaving potassium alone, and the GHK equation collapses back into the Nernst equation for potassium and returns -90-something mV. The resting potential is more positive than potassium’s equilibrium potential precisely because the small but nonzero sodium permeability is constantly leaking a little positive charge inward, dragging the compromise up from where potassium alone would set it.
Here is the conceptual payoff. The resting potential is not the equilibrium potential of any ion. At -70 mV, potassium (equilibrium \sim -90 mV) is not balanced — it has a net force pushing it out. Sodium (equilibrium \sim +60 mV) is enormously out of balance — it has a powerful net force pushing it in, both gradients pointing the same way, since sodium is crowded outside and attracted to the negative inside. Chloride sits close to balance, because its equilibrium potential happens to fall near the resting potential. So the resting neuron is not at peace. It is a dynamic equilibrium: a steady voltage maintained not because the ions have nothing left to do, but because their unsatisfied forces are, for the moment, held in check. Every ion is straining against the membrane. The membrane is a loaded spring.
Try manipulating ion concentrations and permeabilities in the GHK calculator provided below, and observe the changes in the resting membrane potential.
18.4 Keeping the spring loaded: the sodium–potassium pump
If sodium is forever leaking in through the few open sodium channels and potassium forever leaking out through the many open potassium channels, then given enough time, the carefully maintained asymmetry would dissipate, the gradients would flatten, and the tension of our ‘loaded spring’ would dissipate to nothing. Something must continually reload it. That something is a molecular machine: the sodium–potassium pump, also called the sodium–potassium ATPase.
The pump does exactly the opposite of what the leak channels do. Where the leaks let ions slide down their gradients, the pump pushes them up against their gradients, grabbing the sodium that has leaked in and forcing it back out, grabbing the potassium that has leaked out and hauling it back in. On each cycle it ejects three sodium ions and imports two potassium ions. Moving ions against their electrochemical gradients is uphill work, and uphill work costs energy. The pump pays for it by spending one molecule of ATP, the cell’s energy currency, per cycle — which is why it is called an ATPase. The water-tower image from the overview captures the asymmetry exactly: water runs down out of the tower on its own, freely, the way ions slide down their gradients; but getting water back up into the tower takes a pump burning energy, the way restoring the gradients takes the sodium–potassium pump burning ATP.
This single machine is most of the reason the brain is so metabolically expensive. We noted in the overview that the human brain is about two percent of body weight yet consumes roughly twenty percent of the body’s energy at rest, and that a large share of that goes to one molecular machine doing nothing glamorous. This is the machine. The brain spends something on the order of a fifth of your caloric intake, much of it, simply running these pumps to keep the membranes of billions of neurons poised to fire. The fast end of signaling is fast precisely because it is expensive, and the pump is where the energy bill comes due.
Two facts underline how fundamental, and how vulnerable, this machinery is. The pump is not a neuronal invention — every animal cell has it, from sponges and jellyfish to insects and humans, because every animal cell faces the osmotic problem the overview described and solves it the same active way. And because the pump is indispensable, poisoning it is lethal: ouabain, a compound once used on poison arrowheads, works by blocking the sodium–potassium pump, and an animal whose pumps stop will die. (The sodium channel has its own famous poison, tetrodotoxin, the toxin of the pufferfish prepared as the delicacy fugu, which we will meet again when we discuss the channels of the action potential.) The machinery the neuron exploits for thought is the same machinery the cell needs merely to stay alive — and tampering with it kills.
Let us take stock before disturbing the system, because the setup is the whole game:
- There is an asymmetry of ions across the membrane: much potassium inside, much sodium outside, maintained by the pump.
- The membrane is selectively permeable, with many open potassium leak channels and few open sodium channels, so potassium dominates the resting voltage.
- Each ion has its own equilibrium potential, and they differ, so at the resting potential every ion has an unsatisfied driving force.
- The result is a steady but strained resting potential near -70 mV — a loaded spring, held in check, waiting for a release.
A system this poised needs only a small nudge to do something dramatic. The rest of the chapter is about the nudges.
18.5 The triggers: gated channels
What can change the membrane potential? Look back at the GHK equation: the voltage depends on the permeabilities P. Hold the ion concentrations fixed—they barely budge during a single signaling event. The way to move the membrane potential is to change a permeability by opening or closing channels for a particular ion. Open a route for sodium and the membrane, suddenly more permeable to an ion straining to enter, moves toward sodium’s positive equilibrium potential: it depolarizes, becoming less negative inside. Open a route for chloride and the membrane moves toward chloride’s equilibrium potential, typically hyperpolarizing or stabilizing it. Permeability is therefore a key determinant of a neuron’s excitability, and neurons manipulate it by opening and closing channels selective for different ions.
The always-open leak channels we have already met set the resting baseline and do not respond to the moment-to-moment signals considered here. More dynamic are the gated channels, which open or close in response to particular cues. They help make a neuron excitable—capable not merely of maintaining a resting potential, as every cell does, but of changing it sharply in response to a signal. What distinguishes these channels is the trigger that controls them. Together with leak channels, they form four membrane mechanisms to know. (We will also encounter additional ways to open channels in our unit of sensation).
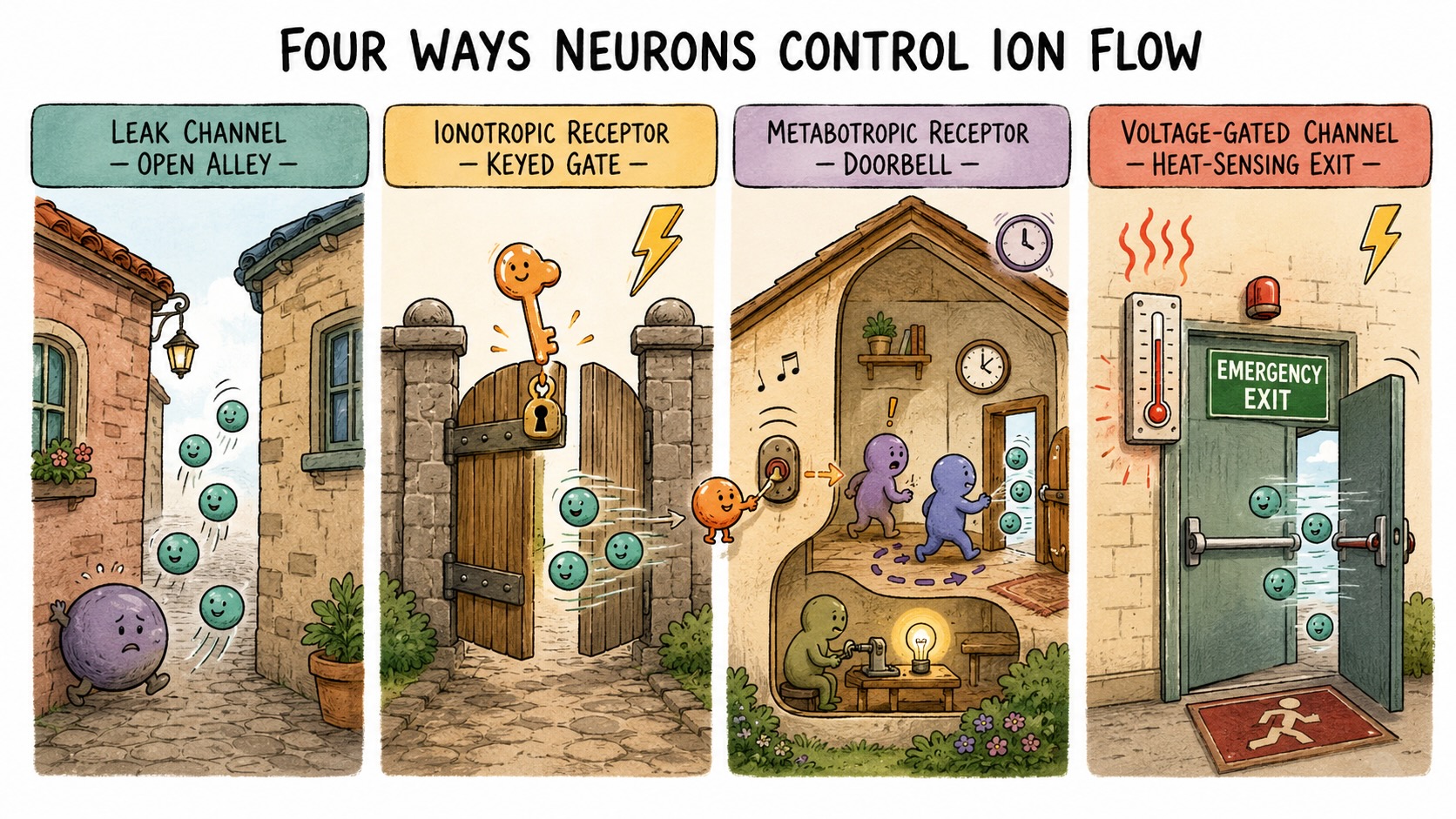
Figure 18.2 uses architectural analogies for these different channels and receptions. A leak channel is an open alleyway between two buildings — no gate at all, nothing deciding whether to admit anyone, though its width imposes selectivity by determining who can fit through. Ions of the right size pass freely in either direction, always. The proportion of leak channels that admit particular ions determines the permeability of the membrane to that ion species and hence determines the resting membrane.
An ionotropic receptor is that same alleyway with a locked gate across it, and the signaling molecule is the key. When the molecule — a neurotransmitter — binds the receptor, the gate swings open and ions pass directly through. Because binding is opening, the response is nearly immediate: a permeability change, and so a voltage change, within a millisecond or two. The full name is a ligand-gated ion channel (“ligand” meaning the molecule that binds), and the speed is its signature. These are the channels of fast, precise, point-to-point synaptic transmission. There are relatively few neurotransmitters that act this way — glutamate, GABA, acetylcholine, serotonin among them — and we will lean on the first two, glutamate and GABA, throughout.
A metabotropic receptor is a doorbell. The signaling molecule presses it from outside, but nothing passes through the wall directly; instead the binding sets off machinery inside the cell, and that internal machinery decides what happens next — perhaps opening a channel elsewhere on the membrane, perhaps doing something that never opens a channel at all. The indirection takes time, so metabotropic effects are slower (tens to hundreds of milliseconds) and can outlast the signal that triggered them. We met cortisol’s surface receptor in the overview as one example of this kind of receptor in Figure 16.1. While certainly relevant to neuronal excitability, we will defer additional discussion of metabotropic receptors and slower time course signaling until Chapter 19.
The fourth type has no ligand at all, and it is the one that will let us solve the distance problem at the end of the chapter. A voltage-gated channel opens not when a molecule binds it but when the membrane voltage around it crosses a threshold. The visual analogy is an emergency door fitted with a heat sensor: it stays locked under ordinary conditions, but if it detects a fire on one side it springs open on its own, no key required. A voltage-gated channel senses not heat but voltage; it carries charged structures in its walls that physically move when the membrane potential shifts, and past a critical voltage they swing the channel open. These channels are how a neuron can respond to its own electrical state, and that capacity for self-triggering is the secret of active propagation. (A fifth type, the mechanically gated channel, opens when physically stretched or pressed; we will meet it in the sensory chapters, where it lets the skin feel pressure and the ear hear sound. It does not figure in the present story.)
With the triggers in hand, we can finally watch a neuron be excited.
18.6 Post-synaptic potentials: the small, local, graded events
Recall the synapse from the previous chapter: the end of one neuron’s axon (the pre-synaptic side) lies a hair’s breadth from a patch of another neuron’s membrane, often a dendritic spine (the post-synaptic side), separated by the narrow synaptic cleft. When the pre-synaptic neuron is active, it releases neurotransmitter into the cleft; the transmitter diffuses across in a fraction of a millisecond and binds receptors on the post-synaptic membrane, opening gated channels. We can now say precisely what that accomplishes.
Before we do, a word about why this chapter dwells on just two neurotransmitters when the brain is known to use more than a hundred. The reason is that two of them do the overwhelming bulk of fast point-to-point signaling — something we refer to colloquially as wired transmission. These two neurotransmitters are also an opposed pair. Glutamate is the principal excitatory transmitter of the central nervous system and the most abundant neurotransmitter in the brain; by one widely cited estimate it is used at on the order of ninety percent of the synaptic connections in the human brain. GABA (gamma-aminobutyric acid) is its inhibitory counterpart, the principal inhibitory transmitter, present at something like a third of all synapses — more than any transmitter except glutamate. The same opposition shows up if you count neurons rather than synapses: in the cerebral cortex, roughly eighty percent of neurons are excitatory and glutamatergic and roughly twenty percent are inhibitory and GABAergic, a four-to-one ratio that holds with remarkable consistency across mammalian species. (There is a tidy economy in the pairing that hints at why it is so universal: GABA is manufactured directly from glutamate, in a single enzymatic step, so the brain builds its master “off” switch out of its master “on” switch.) Between them, glutamate and GABA are the fast vocabulary of the brain — the molecular form of the excitation and inhibition that the previous chapter called the first grammar of neural signaling. The dozens of other transmitters are not unimportant, but most of them modulate this fast traffic rather than carry it, and they belong to Chapter 19. Here we follow the two that do the carrying.
18.6.1 Excitation: the glutamate synapse
Take the most common case first, an excitatory synapse using glutamate. Glutamate binds ionotropic glutamate receptors on the post-synaptic membrane and opens channels permeable to sodium. And we know what sodium does when given a route: it is crowded outside and attracted to the negative inside, both gradients pointing the same way, so it floods in under a strong electrochemical driving force. The inrush of positive charge makes the local membrane less negative — it depolarizes it, nudging the voltage from -70 mV up toward zero. This local depolarization is a post-synaptic potential, and because it is depolarizing — because it pushes the cell toward activity — it is an excitatory post-synaptic potential, or EPSP.
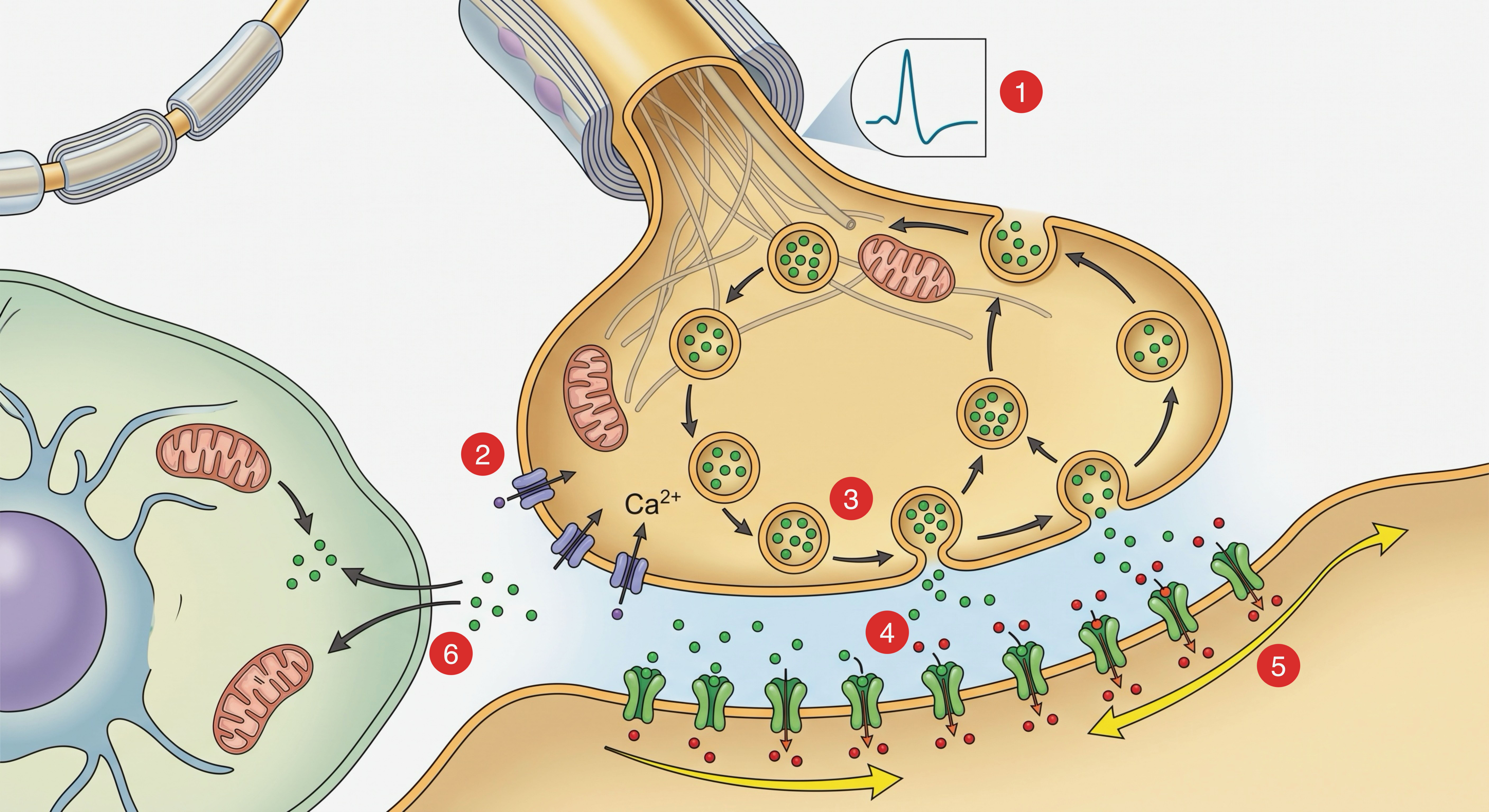
Figure 18.3 shows the sequence of events at an excitatory glutamatergic synapse. A presynaptic terminal, or axon bouton (yellow, upper right), arising from a myelinated axon (yellow and segmented, upper left), forms a synapse with a postsynaptic dendrite (lower right). A closely apposed astrocyte (light green) surrounds part of the synapse and helps regulate its chemical environment.
Arrival of the action potential. An action potential, represented by the blue voltage trace in the upper-right inset, travels along the myelinated axon and reaches the yellow presynaptic bouton. The resulting depolarization provides the electrical signal that initiates neurotransmitter release.
Calcium entry. Depolarization opens voltage-gated calcium channels, shown as purple-blue channels in the presynaptic membrane. Calcium ions (blue spheres) enter the bouton and activate the molecular machinery that controls synaptic-vesicle fusion.
Glutamate release and vesicle recycling. Glutamate-filled vesicles move from reserve pools toward the active zone, dock at the membrane, and fuse with it, releasing glutamate (green spheres) into the synaptic cleft. Vesicular membrane is then retrieved, re-formed into vesicles, refilled with glutamate, and returned to the reserve or ready-to-release pools; mitochondria (orange-red) supply ATP for this cycle.
Activation of postsynaptic receptors. Glutamate diffuses across the cleft and binds to ionotropic glutamate receptor-channels (green) in the postsynaptic membrane. These channels open, allowing positively charged ions—especially sodium (red spheres)—to enter the dendrite.
Generation of an EPSP. The inward flow of positive charge produces an excitatory postsynaptic potential, or EPSP, represented by the thick yellow arrows within the dendrite. This graded potential spreads passively along the dendritic membrane toward the cell body and axon hillock, where it may combine with other postsynaptic potentials.
Astrocytic clearance and recycling. Processes of the astrocyte (light-green) remove glutamate from the synaptic cleft, sharpening the signal and preventing persistent receptor activation and excitotoxicity. The astrocyte, which contains a nucleus (purple) and orange-red mitochondrion (orange-red), converts glutamate to glutamine for return to the presynaptic terminal and also buffers extracellular potassium, making this a tripartite synapse in which glial regulation is essential to reliable transmission.
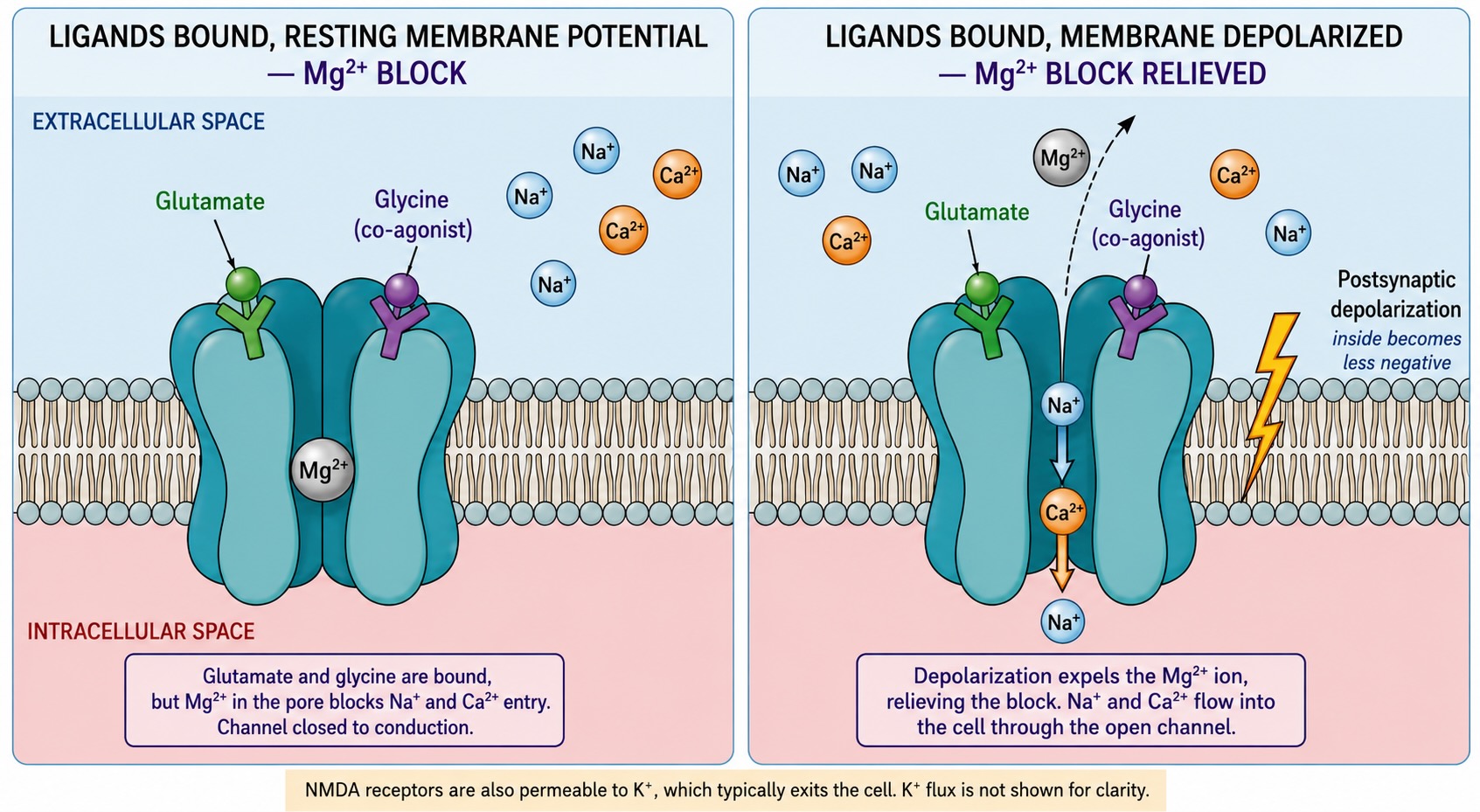
The ionotropic glutamate receptors are not all alike. The AMPA receptor depicted in Figure 18.3 is the workhorse: it opens fast when glutamate binds, admits sodium, and produces the brief EPSP just described. The NMDA receptor looks similar but has a peculiar extra feature — at the resting potential its channel is physically plugged by a magnesium ion, so glutamate alone cannot open it; the plug pops out only when the membrane is already depolarized. This makes the NMDA receptor a detector of two things happening at once, and we will see in Chapter 20 that this coincidence-detecting property is the molecular heart of synaptic learning. (This is the cameo the magnesium row of our ion table was waiting for. Notice that magnesium’s job here has nothing to do with carrying current down a steep gradient — its gradient is nearly flat — and everything to do with sitting in a channel until the voltage tells it to leave. The shallow gradient is the point: magnesium is a plug, not a signal.) Glutamate also binds slow metabotropic receptors, the doorbell type, on the post-synaptic neuron and on the surrounding astrocyte; those act over a longer timescale and we set them aside for now, as our focus in this chapter is on the fast, millisecond timebase signaling.
In the overview to this unit, I said the meaning of a signaling molecule is not carried by the molecule but decided by the receptor it meets. Glutamate is the cleanest proof. One and the same glutamate molecule produces a fast brief depolarization at an AMPA receptor, opens the coincidence-detecting NMDA receptor only when the membrane depolarization is right, or sets off a slow internal cascade at a metabotropic receptor lasting hundreds of milliseconds. One molecule, several receptors, several different consequences on several different timescales. The transmitter is not the message; the receptor is.
18.6.2 Inhibition: the GABA synapse
Now the inhibitory case. When an inhibitory neuron fires, it releases GABA onto the post-synaptic membrane, and GABA — like glutamate — has both a fast ionotropic receptor and a slow metabotropic one. The fast one, the GABA-A receptor, is an ionotropic channel permeable not to sodium but to chloride, and here the two-gradient logic plays out differently than it did for sodium. As we shall see, inhibition is gentler and more conditional than excitation.
Look back at the ion table. Chloride is more concentrated outside the cell, so its concentration gradient points inward; but chloride is negative and the inside of the cell is already negative, so its electrical gradient points outward. The two forces very nearly cancel — which is exactly why chloride’s equilibrium potential falls so close to the resting potential, and why its net driving force is weak. When a GABA-A channel opens with the membrane near rest, the small net movement of negative charge tends inward, making the inside slightly more negative, pushing the voltage away from zero and away from the firing threshold. This hyperpolarizing change is an inhibitory post-synaptic potential, or IPSP — the mirror image, in sign, of the EPSP.
But the cancellation of chloride’s two gradients has a subtler consequence. Because chloride’s equilibrium potential sits so near the resting potential, the direction chloride moves depends on the exact local voltage at the moment the channel opens. If the membrane has been depolarized by nearby excitation, chloride flows inward and pulls the voltage back down; if the membrane is already at or below rest, chloride may barely move at all, or even flow outward. Either way, the open chloride channel does something beyond shifting the voltage: it makes the membrane leaky. An open channel is a hole, and a hole lets charge escape. So when an excitatory current arrives at a patch of membrane riddled with open GABA-A channels, much of that current leaks straight back out through the chloride holes instead of spreading toward the axon — the excitation is bled away before it can do its work. This is called shunting inhibition, and it is often more important than hyperpolarization itself. It is also why the placement of inhibitory synapses matters so much: an inhibitory synapse sitting near the axon hillock, or on the cell body, or even on the axon’s initial segment — recall the chandelier and basket cells of the previous chapter, positioned at exactly these points — can shunt away the summed excitation of the entire dendritic tree just as it is about to trigger a spike. Inhibition placed at a chokepoint is worth far more than its small voltage change suggests.
GABA’s slower receptor, the metabotropic GABA-B receptor, works by the doorbell mechanism rather than by opening a channel directly. Its internal cascade opens potassium channels, and potassium — which, as the table shows, is concentrated inside and has an outward gradient — flows out, carrying positive charge out of the cell and hyperpolarizing it over a longer, slower time course (hundreds of milliseconds). So GABA inhibits twice over: a fast chloride-based IPSP through GABA-A, and a slow potassium-based hyperpolarization through GABA-B.
The contrast between GABA and glutamate ionotropic channels is important to remember, because it is the molecular root of an asymmetry that runs through all of neural computation. Glutamate opens a sodium channel onto an ion straining to enter, and excitation arrives hard, fast and unconditionally — sodium always rushes in. GABA opens a chloride channel onto an ion sitting nearly at equilibrium, and inhibition arrives gently and conditionally, doing much of its work not by driving the voltage down but by holding it near rest and shunting excitation away. Excitation and inhibition are opposite in sign, but they are not simply mirror images; they have different characters, and those characters fall directly out of where each ion’s two gradients happen to leave it.
18.6.3 Why a local event spreads, and why it fades
A post-synaptic potential begins as a strictly local affair — sodium enters at one small patch of membrane, on one spine — yet its job is to influence the distant axon hillock, where the decision to fire is made. How does a local voltage change reach a faraway place? And, just as important, why does it weaken on the way?
It spreads because the charge that enters does not stay put. Sodium ions that rush in are positive, and like charges repel; they push away from one another and from the other positive ions nearby, so the positivity diffuses along the inside of the membrane, away from the point of entry. The depolarization, in other words, propagates sideways from the synapse, a spreading wave of “less negative.”
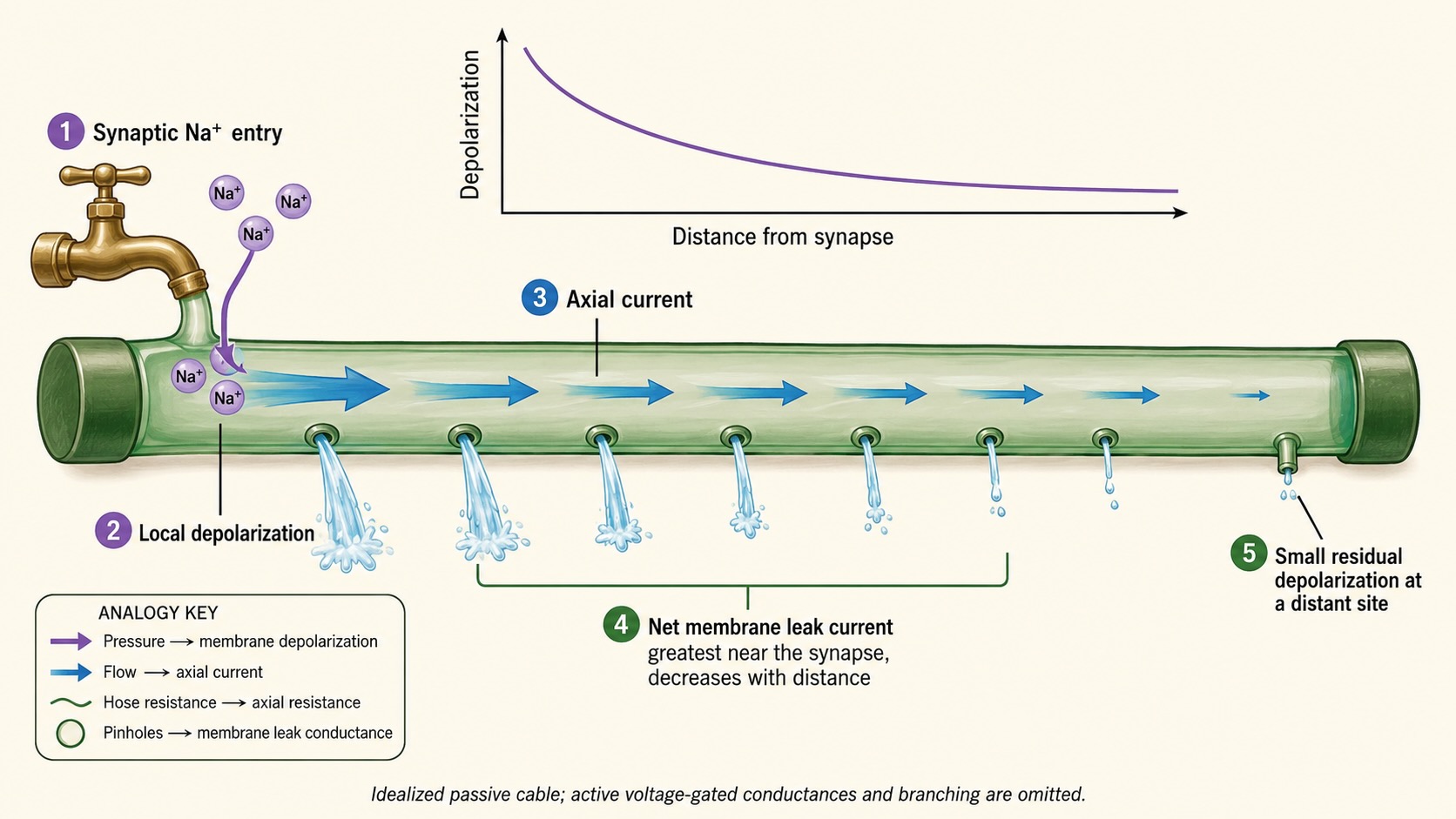
The neuron is a closed system governed by the simple conservation of charge: charge that enters at one location must be balanced by charge that leaves elsewhere. As a wave of positive depolarization spreads internally from a synapse, it forces potassium out through the membrane’s ever-present leak channels, bleeding away the signal as it travels. Picture an irrigation hose with regularly spaced holes along its length and a capped far end: turn on the tap briefly, and the holes nearest the source spray the hardest, while those at the far end barely dribble. Yet, no matter how long the hose, a little water always reaches the end. The post-synaptic potential behaves just like this—short-lived because the synaptic channels quickly close, and graded because it falls off smoothly with distance rather than failing all at once. This passive, decremental spread is called electrotonic conduction, and its defining feature is that nothing regenerates the signal along the way. Because it is all downhill from the synapse, a single EPSP originating at a distant spine will reach the axon as a mere whisper, and to matter, those whispers must combine.
18.7 Summation: the neuron as an analog computer
A real neuron may carry hundreds or thousands of synapses scattered over its dendrites and soma, many of them active at once, each launching its own little graded wave toward the axon. The place where these waves converge is the axon hillock (also called the initial segment or trigger zone), the specialized patch where the axon leaves the soma. The hillock experiences the running total of all those arriving potentials — the EPSPs as positive contributions, the IPSPs as negative ones — and it is this continuous adding-up that constitutes the neuron’s information processing. The neuron is, in this sense, an analog computer: it sums graded inputs into a graded result.
Return to the garden hose, but now imagine many hoses, all their capped far ends resting on the same small patch of grass. Pulse water through all of them at once and that patch receives a little from each; the total is the sum of the dribbles, and with enough hoses the total can be substantial. The axon hillock is that patch of grass. It registers the sum of every PSP the neuron is currently experiencing. Three things shape that sum, and each is worth stating plainly because each becomes a lever the brain can pull.
The first is sign: EPSPs and IPSPs add as positive and negative numbers. A neuron receiving strong excitation and strong inhibition at the same time may sum to nothing at all — the inhibition cancels the excitation — which is exactly what makes inhibition useful. The grammar of excitation and inhibition from the previous chapter is, at the membrane, literally arithmetic.
The second is space — spatial summation. Because every PSP decays with distance, a synapse near the axon hillock delivers more of its punch than an identical synapse far out on a distal dendrite. Location is leverage. (This has a striking consequence: an excitatory synapse and an inhibitory synapse placed near each other can annul one another, while the same two synapses placed at different distances from the hillock can let the excitation win. The brain exploits exactly this geometry, positioning particular inhibitory synapses — recall the chandelier and basket cells of the previous chapter — at points of maximal leverage.)
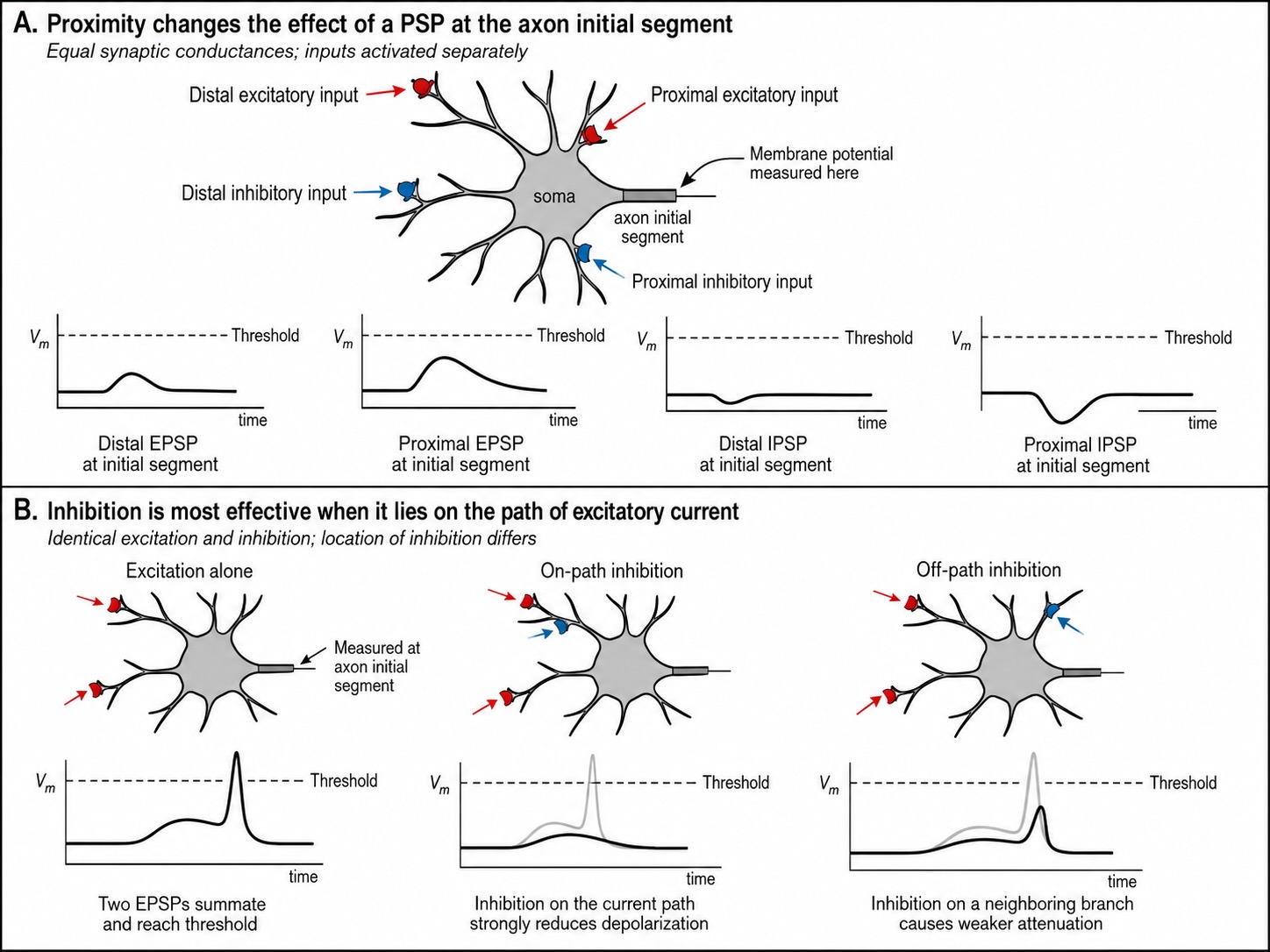
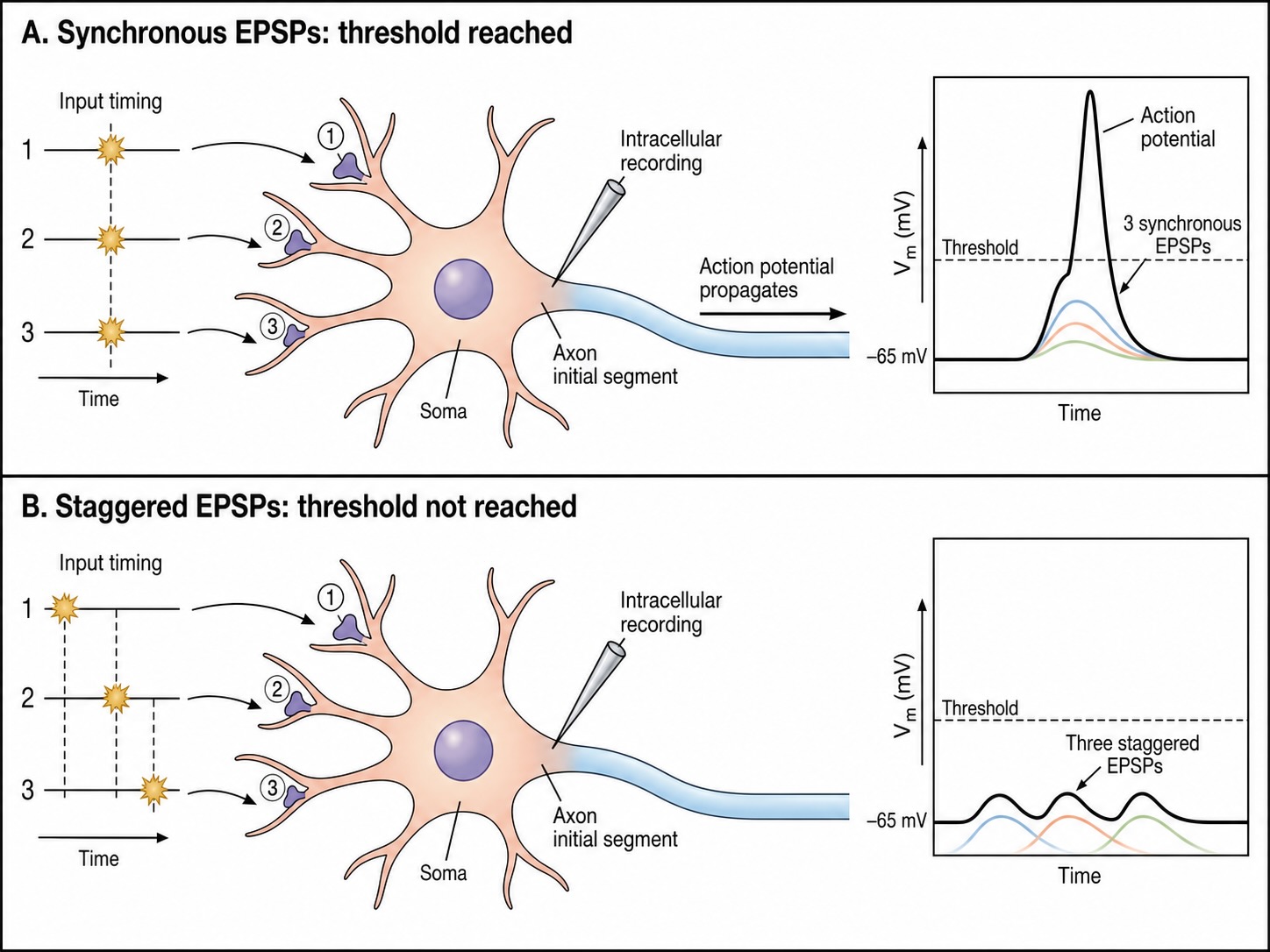
The third is time — temporal summation. Each PSP has a brief duration, and PSPs that arrive close together in time overlap and add, while those spread too far apart decay between arrivals and never accumulate. Synchrony is therefore powerful: inputs that fire together sum together. The lecturer’s classroom demonstration captures it — if a room full of people clap at the same instant the sound is loud, but if they clap one after another it is merely a patter, even though the same number of claps occurred. The same total input, concentrated in time, has a far larger effect.
Putting it together: the axon hillock computes a spatiotemporal sum over dozens to thousands of synapses, weighting each by where it sits and when it fires, treating excitation and inhibition as opposite signs. If that sum depolarizes the hillock past a critical threshold — typically around -50 mV — something new and qualitatively different happens. Up to threshold, everything has been graded and passive and local. At threshold, the neuron stops summing and starts shouting.
The garden-hose analogy hides some real and consequential physics. How far a post-synaptic potential spreads before fading depends on the cable properties of the dendrite — the same mathematics that engineers use to describe signal loss in undersea telegraph cables, imported into neuroscience by Wilfrid Rall. The decisive variable is the dendrite’s diameter: a thick dendrite carries a potential farther, and faster, than a thin one, because a wider cable offers the internal current less resistance. Branching pattern matters too, and so does a subtler structural feature — the dendritic spine itself, whose narrow neck can electrically amplify the local potential it receives. Two synapses delivering identical input can therefore have very different effects depending on the caliber, branching, and spine geometry of the dendrite that carries them. The “analog computer” is not a uniform summing device; it is shaped by its own anatomy.
There is a further wrinkle that complicates the tidy picture of passive dendrites and active axons. The standard textbook story says voltage-gated channels are located in the axon and the dendrites merely conduct passively. But voltage-gated channels turn out to inhabit some dendrites as well, and where they do, a distant dendritic region can generate its own regenerating event — a dendritic spike — that carries a far-flung input toward the soma without the usual decay. Imagine a synapse stranded at the end of a long apical dendrite, ordinarily too remote to matter; if its branch can mount a dendritic spike, it suddenly has an express line to the hillock instead of a slow leak. This effectively turns parts of the dendritic tree into semi-independent processing units, each integrating its local inputs and then forwarding the result actively. It is one of several ways, foreshadowed in the previous chapter’s discussion of where the neuron doctrine fails, that real neurons are richer than the simple model of passive dendrites and a single trigger zone.
18.8 The action potential: the large, regenerating, all-or-none event
We have reached the problem the whole chapter has been building toward. Post-synaptic potentials decay with distance; over a millimeter of dendrite they fade to a whisper. Yet a neuron may need to send its output a very long way — from your spinal cord to your foot, or, in a giraffe, down an axon some fifteen feet long. A signal that dies out over a millimeter cannot possibly cross a meter by passive spread. Passive conduction, by its nature, only loses. To carry a signal over distance, the signal must be actively regenerated — rebuilt to full strength again and again along the way, so that what arrives at the far end is as strong as what left.
The means of regeneration is the third kind of gated channel, the voltage-gated channel, deployed in a particular arrangement. The axon hillock and the axon are packed with voltage-gated sodium channels, and these are the emergency doors that open in response to voltage. Here is the sequence, the event called the action potential, and it is worth following ion by ion because every step is a consequence of the gradients we have already established.
The summed PSPs depolarize the axon hillock toward threshold. At about -50 mV, the voltage-gated sodium channels there sense the change and snap open. Now sodium has a wide-open route, and we know what sodium does with a route — it floods in, driven hard by both its gradients. This inrush depolarizes the membrane further, which opens still more voltage-gated sodium channels, which admits more sodium, which depolarizes further still: a self-reinforcing avalanche. The membrane potential does not drift gently upward; it rockets toward and past zero, overshooting until the inside is briefly positive — around +30 mV — relative to the outside. This is the upstroke of the spike, sodium’s loaded spring discharging all at once.
Now comes the switchover. I will be precise about it because the exact sequence gives the spike its shape. As the membrane climbs toward +30 mV, two things happen in close succession, and together they end the upstroke and begin the downstroke. First, the voltage-gated sodium channels inactivate. Inactivation is not the same as simply closing: a separate part of the channel — often pictured as a hinged plug on the channel’s inner mouth — swings shut and corks it, and the channel cannot reopen until the membrane has repolarized and the plug has been reset. This is a one-way valve slamming, and it cuts off the sodium inrush abruptly. (Note the timing: sodium’s own equilibrium potential is about +60 mV, so the channels inactivate while sodium would, given the chance, still keep pouring in. The spike is self-limiting; it shuts itself off well before the sodium gradient is exhausted.) Second, at almost the same high voltage, the slower voltage-gated potassium channels — which sensed the same depolarization the sodium channels did, but respond with a built-in delay — finally swing open. Potassium is concentrated inside and is now also repelled by the freshly positive interior, so both of its gradients point outward at once, and it rushes out, carrying positive charge with it.
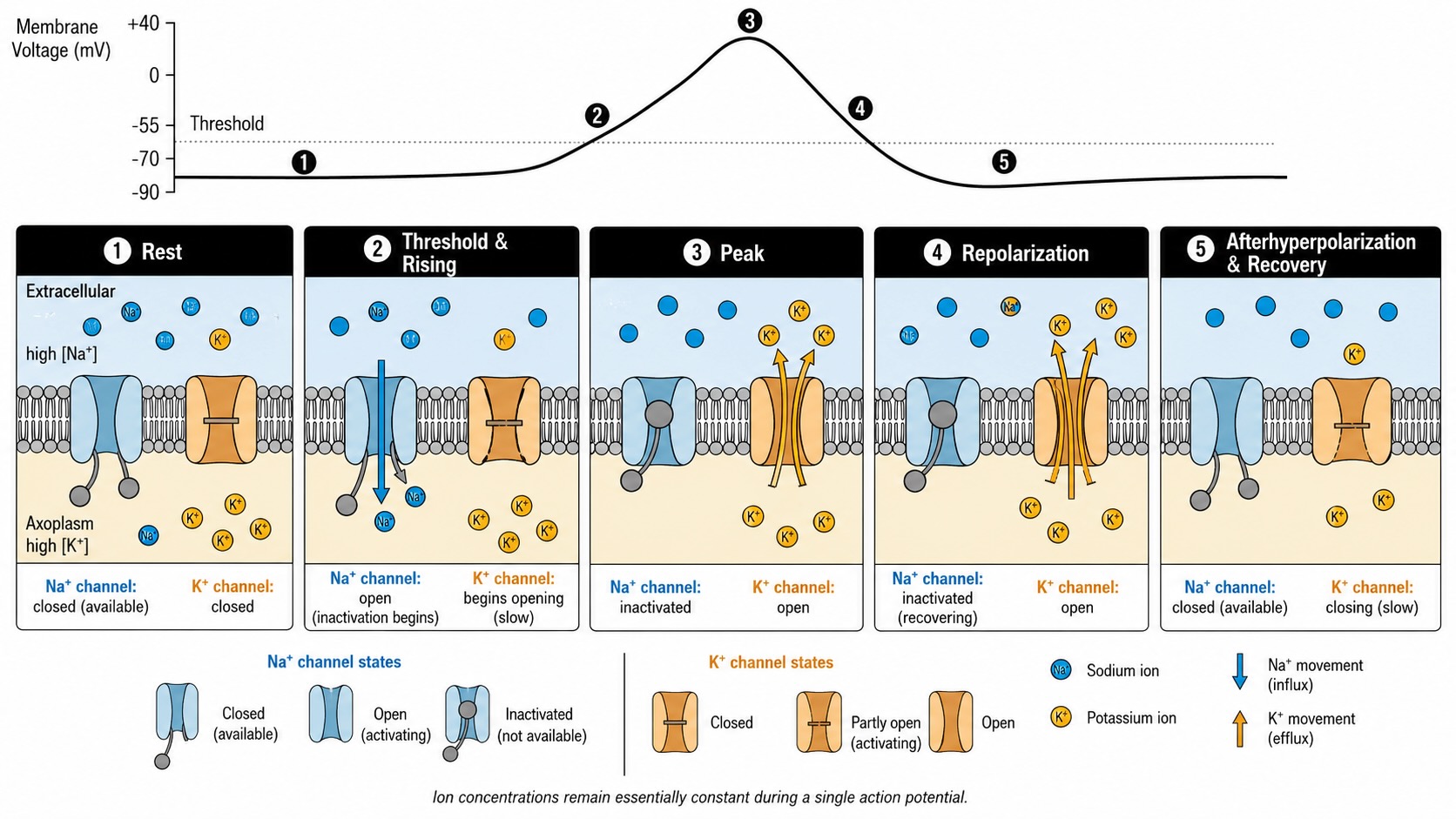
So the changeover is a relay: sodium channels open fast and inactivate at the peak; potassium channels open slow and take over just as sodium quits. The outrush of potassium drives the membrane potential back down — the downstroke, or repolarization. And because the potassium channels are sluggish to close, potassium keeps leaving a moment too long, so the membrane overshoots its resting value and dips briefly more negative than rest, down toward -80 mV, a phase called the afterhyperpolarization. The whole event — rest to +30 mV and back below rest — takes only about a millisecond or two.
I will emphasize two features of this event because they distinguish the action potential sharply from the post-synaptic potentials that preceded it. First, it is all-or-none. Below threshold, nothing happens; at threshold, the full avalanche fires; and a stronger-than-threshold stimulus produces no bigger a spike — the action potential is the same size no matter how far past threshold the trigger went. There is no graded, partial action potential. The neuron’s analog summation feeds into a digital decision. Second, and consequently, it is self-propagating. When a patch of axon fires, the sodium that floods in depolarizes the neighboring patch past its threshold, opening its voltage-gated channels, which depolarizes the next patch, and so on — voltage-gated channels falling open in sequence like a line of dominoes, each one knocking over the next. Because every patch rebuilds the spike to full amplitude, the signal that reaches the end of a meter-long axon is exactly as strong as the one that left the hillock. The decay problem is solved, not by preventing decay, but by regenerating the signal faster than it can decay.
Now account for what the spike has left behind, because this is where the metabolic bill and the limits on firing both come from. During the upstroke, sodium poured in; during the downstroke, potassium poured out. The net result, in the thin shell of axon just under the membrane where the action potential happened, is that the two ions have partly traded places: there is now excess sodium inside and excess potassium outside — the local concentrations have been nudged toward reversal by the very event that exploited them. (The amounts are small relative to the total ion content, which is why a single spike barely dents the gradients and a neuron can fire many times before exhaustion; but the direction of the change is exactly opposite to the resting arrangement the cell works to maintain.) Restoring the resting distribution is not something the gradients can do on their own — it means pushing sodium back out and potassium back in, against their gradients, which is uphill work. This is the job of the sodium–potassium pump, grinding away on ATP, three sodium out and two potassium in per cycle, reloading the spring for next time. The action potential spends the gradient; the pump restores it; and the brain’s enormous resting energy budget is, in the end, largely the bill for running this cycle across billions of neurons firing again and again.

This cleanup requirement has a direct and important consequence: a neuron cannot fire arbitrarily fast, and the action potential is therefore a discrete, countable event rather than a continuous one. The limit is called the refractory period, and it comes in two stages. Immediately after a spike, while the sodium channels are still inactivated — corked by that inner plug, which resets only once the membrane has repolarized — the neuron simply cannot fire again, no matter how strong the input. This is the absolute refractory period, and it lasts about one to two milliseconds. It sets a hard ceiling on firing rate (a neuron with a one-millisecond absolute refractory period cannot exceed roughly a thousand spikes per second), and it does something else useful: because the patch of membrane just behind an advancing spike is absolutely refractory, the spike cannot back-propagate along the axon into territory it has already crossed, so the action potential is forced to travel in one direction only, from hillock to terminal. After the absolute period comes a relative refractory period, during which the afterhyperpolarization still has the membrane below its resting value and the sodium channels are only partway reset; here the neuron can fire, but only in response to a stronger-than-usual input, because it is starting from farther below threshold. Together these two phases ensure that spikes stay separated in time, that there is a ceiling on how fast a neuron can signal, and that the wave runs cleanly in one direction down the axon.
Notice what the action potential is not. It is not a new kind of energy, and it is not fundamentally different machinery from the post-synaptic potential. It is the same business — ions moving down their electrochemical gradients through channels — with one organizational difference: the channels are voltage-gated and arranged in a line, so that each patch’s discharge triggers the next. The action potential is the controlled release of the loaded spring the pump spent all that energy to load. We did not need to invent a new force for long-distance signaling. We needed only to arrange the old one to regenerate itself.
There is one more consequence of the spike to flag. I have just said that the action potential cannot run backward along the axon, because the membrane behind it is refractory. But the axon is not the only direction it can go. The spike is born at the axon hillock, and from there it does not only travel outward down the axon — a wave of depolarization also spreads backward, from the hillock into the soma and up into the dendritic tree. This is the back-propagating action potential. In part it spreads passively, the way any depolarization spreads through the cell; and in many neurons the dendrites carry their own voltage-gated channels (the dendritic spikes of an earlier deeper dive), which let the back-propagating signal travel into the dendrites with less decay than passive spread alone would allow. Either way, the result is the same and it is remarkable: when a neuron fires, every one of its dendritic spines briefly learns that it fired. A wave of depolarization washes back over the synapses that fed the cell, arriving after they did their work, carrying the news that their collective effort succeeded in producing an output.
Why should that matter? Because it is precisely the signal a synapse would need in order to learn. A synapse that wants to strengthen itself only when it genuinely contributes to making the neuron fire needs two pieces of information: that it was active, and that the neuron fired. The first it knows locally, from the glutamate in its own cleft. The second is exactly what the back-propagating action potential delivers — a cell-wide announcement of the output, reaching back to coincide, at the synapses that were recently active, with the trace of their own activity. We met the molecular detector built for exactly this coincidence a few pages ago: the NMDA receptor, glutamate-bound but magnesium-plugged, waiting for a depolarization to pop the plug. The back-propagating spike is one source of that depolarization. We will not develop this now, but the architecture is worth seeing in advance: the fast machinery of this chapter does not only carry information forward to the next neuron, it also feeds a copy of its own output backward to the synapses that produced it — and that backward copy is the raw material from which, in the final chapter of this unit, synapses will build memory.
The account above is qualitative — sodium in, potassium out, threshold and overshoot and refractoriness. The quantitative theory, one of the great achievements of twentieth-century biology, came from Alan Hodgkin and Andrew Huxley, working at Cambridge with an unusually convenient piece of nature: the giant axon of the squid, an axon so thick (up to a millimeter across) that an electrode could be threaded down its length. Their collaboration was interrupted by the Second World War and resumed after it; in 1952 they published a series of five papers that reconstructed the action potential from measurements of how the membrane’s conductance to sodium and potassium changes with voltage and time. They received the Nobel Prize for it in 1963.
Their central experimental tool was the voltage clamp, a feedback circuit that holds the membrane at a chosen voltage while measuring the current required to keep it there — which is, in effect, a direct readout of how much the channels are open. By clamping the axon at different voltages and watching the currents, Hodgkin and Huxley could separate the sodium current from the potassium current and characterize each one’s dependence on voltage and time. They found that the sodium conductance rises fast and then inactivates, while the potassium conductance rises more slowly and persists — exactly the two processes that, in the qualitative story, produce the upstroke and the downstroke.
They then captured this in a set of differential equations — the Hodgkin–Huxley model — in which the total membrane current is the sum of a capacitive current and the ionic currents, each ionic current being a voltage- and time-dependent conductance multiplied by the driving force on that ion. Written compactly, the membrane obeys
C_m \frac{dV}{dt} = -\left[ g_{\text{Na}}(V - E_{\text{Na}}) + g_{\text{K}}(V - E_{\text{K}}) + g_{\text{L}}(V - E_{\text{L}}) \right] + I_{\text{ext}}
where each E is the equilibrium potential we learned to compute with the Nernst equation, and each conductance g rises and falls according to its own auxiliary equations governing the channels’ opening and closing. Notice that the driving force on each ion is written (V - E_{\text{ion}}) — the difference between the present membrane voltage and that ion’s equilibrium potential — which is precisely the two-gradient idea from the start of the chapter, now in symbols: an ion’s push is proportional to how far the membrane is from the voltage that would balance it. The whole chapter, in a sense, is a long verbal unpacking of that one parenthesis. Solving the equations reproduces the action potential’s shape, its threshold, its all-or-none character, its speed, and its refractory period, from channel behavior alone. It was the first time a complex piece of physiology had been derived, quantitatively, from the properties of its molecular parts, and it set the template for computational neuroscience ever after.
18.9 Speed: myelin and saltatory conduction
The domino model regenerates the signal at full strength, but it does not, by itself, make the signal fast. Each voltage-gated channel takes a little time to sense its neighbor’s depolarization and open, and if every microscopic patch of membrane must take its turn in sequence, conduction down a long axon is reliable but slow. Evolution’s solution to the speed problem reuses a cell we met in the previous chapter.
The oligodendrocyte, a type of glial cell, wraps lengths of axon in myelin, a fatty insulating sheath. (In the peripheral nervous system the wrapping is done by a different glial cell, the Schwann cell, but the principle is identical.) The myelin is not continuous: it is laid down in segments separated by small bare gaps called the nodes of Ranvier, where the axon membrane — and its voltage-gated sodium channels — is exposed. The effect on conduction is dramatic, and the garden hose explains why. Picture again a hose leaking from holes along its length; the pressure bleeds away as it travels. Now wrap most of the hose in tape, leaving only occasional gaps unwrapped. The water can no longer leak out through the taped stretches, so it is forced down the inside of the hose, arriving at the next open gap with nearly undiminished pressure. Myelin is the tape. Between nodes, the insulated membrane cannot leak current, so the depolarization is driven down the inside of the axon with little loss, and it only needs to be actively regenerated at the nodes, where the sodium channels sit. The action potential thus appears to jump from node to node rather than crawling continuously — a mode of conduction called saltatory, from the Latin for “leaping.”
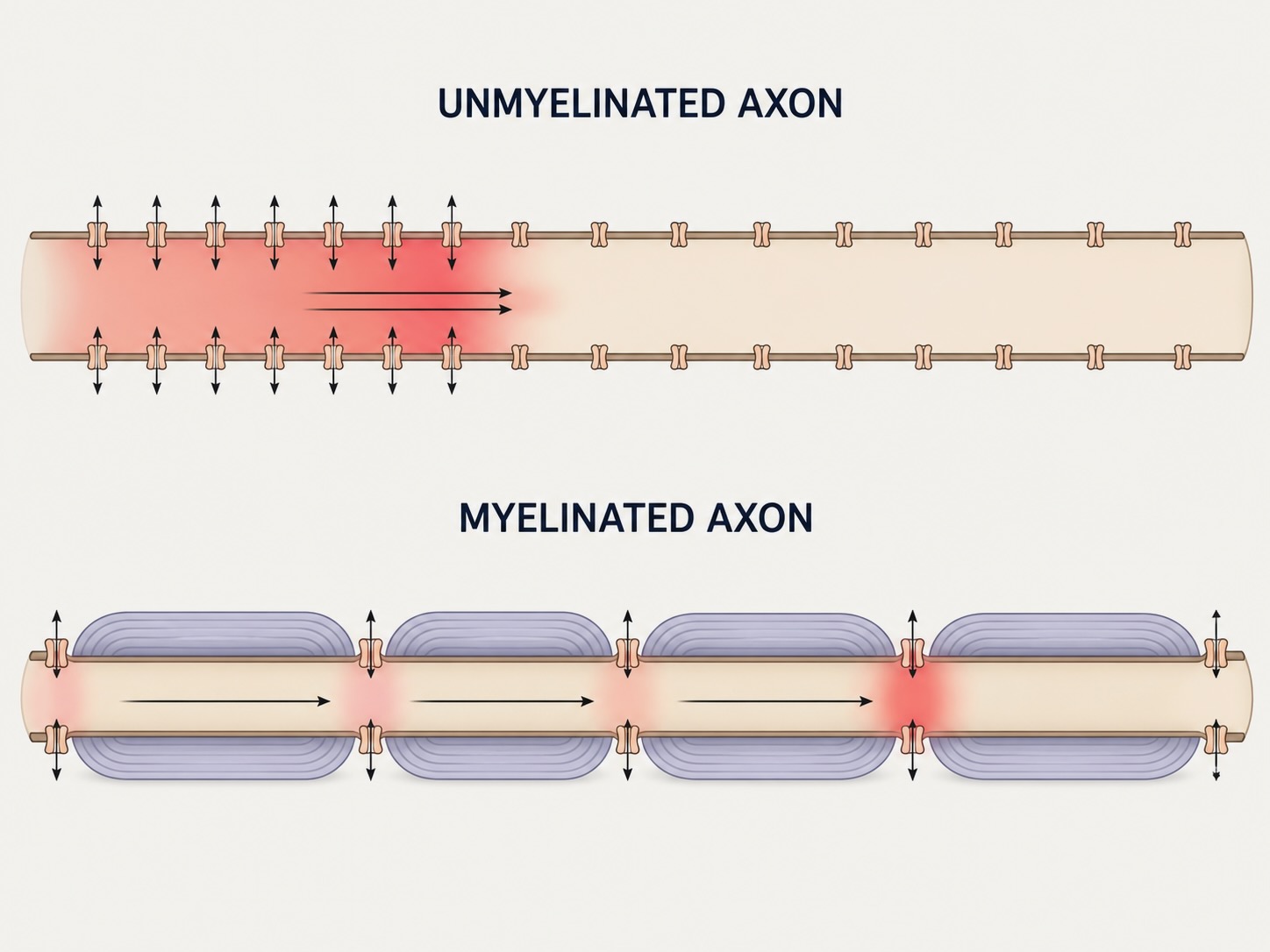
The payoff is large. An unmyelinated axon conducts at perhaps 20 meters per second; a myelinated one can exceed 80. For a signal that must travel from the spinal cord to the brain, or from a fingertip to the cord when you touch something hot, that difference is the difference between a reflex quick enough to protect you and one too slow to matter. (Axon diameter matters too, by the same cable logic as in the dendrites: thicker axons conduct faster. Myelin and caliber together set a neuron’s conduction speed.) The importance of myelin is thrown into relief by what happens when it fails: in multiple sclerosis and other demyelinating diseases, the insulating sheath is attacked and stripped away, the bare internodal membrane leaks current it was never built to handle, the gaps between functioning nodes grow too wide for passive spread to bridge, and conduction slows, stutters, or fails outright — producing the motor and sensory disturbances of the disease. Myelination is also slow to finish in development, continuing into adolescence in some brain regions.
18.10 Where this leaves us
We set out to explain how a neuron is excited, and we can now state the answer compactly. Every fast electrical event in a neuron represents the outcome of two gradients — chemical and electrical — acting on the same ions, and about channels that decide, moment to moment, which ions may move. The pump spends energy to hold the membrane in a disequilibrium, a ‘spring-loaded’ state, far from equilibrium for every ion at once. Gated channels release that strain selectively: a glutamate-opened sodium channel produces a small local depolarization, an EPSP; a GABA-opened chloride channel produces a small hyperpolarization, an IPSP. These graded potentials spread passively and fade, but a neuron sums thousands of them across space and time at its axon hillock, treating excitation and inhibition as opposite signs — analog computation. If that sum crosses threshold, voltage-gated channels arranged in a line fire in sequence, regenerating an all-or-none action potential that carries the result, undiminished, down an axon of any length, with myelin to make it fast. And then the pump cleans up, on ATP, and the bill for all of it is much of the brain’s metabolic cost.
Three threads from the unit have run through this chapter. The two-gradient idea is the chapter’s spine, and it reappeared in the Hodgkin–Huxley driving force (V - E_{\text{ion}}) as the formal statement of everything I said in words. The molecule-versus-receptor principle from the overview found its sharpest proof in glutamate, whose meaning — fast or slow, brief or lasting — is set entirely by the receptor it meets. And the astrocyte from the previous chapter turned out to be a working participant in fast signaling, not a bystander, clearing the glutamate that would otherwise poison the synapse and buffering the potassium that signaling sheds.
Two large concepts have been deliberately set aside, and the next chapters take them up. In Chapter 20, we will consider how a synapse’s strength changes with use — how the weight of a connection is turned up or down, which is the cellular basis of learning and memory. We have, in fact, quietly laid its foundations in this chapter: the NMDA receptor that opens only when activity and depolarization coincide, and the back-propagating action potential that carries word of the neuron’s output back to the synapses that caused it. Those two pieces are the raw material of synaptic plasticity, and the final chapter of this unit assembles them into a mechanism for memory. The second is the slower, metabotropic mode of signaling we have only gestured at — the doorbell that sets off internal cascades rather than opening a channel outright. That mode is the gateway to neuromodulation and to the chemical anatomy of the brain: the diffuse systems that bathe whole populations of neurons in serotonin, dopamine, noradrenaline, and acetylcholine, shifting their excitability over seconds and longer rather than carrying information point to point. Having built the fast electrical layer from first principles, we now widen the frame to the slower chemical environment in which all of that fast signaling is bathed.